
Introduction
Sterile packaging is the last line of defense between a medical product and contamination. For businesses distributing or selling medical products, understanding it is essential for compliance and patient safety.
The stakes are measurable. According to the CDC, surgical site infections (SSIs) account for 20% of all healthcare-associated infections, with approximately 110,800 inpatient SSIs occurring in 2015 alone. These infections extend hospital stays by 9.7 days and cost the healthcare system roughly $3.3 billion annually. Compromised sterile packaging directly contributes to these preventable events.
This article covers what sterile packaging is, the types and materials used, regulatory requirements, and the key considerations every medical product business should know to protect patients and maintain compliance.
TLDR:
- Sterile packaging creates a microbial barrier that prevents contamination from manufacture through use
- ISO 11607 and FDA regulations govern packaging materials, validation, and sterilization processes
- Packaging failures trigger costly recalls, regulatory action, and patient harm
- Material choice must match sterilization method; incompatible combinations cause failure
- Storage, handling, and logistics partners must maintain FDA, ISO, and GMP compliance
What Is Sterile Packaging?
Sterile packaging is the process of enclosing medical devices or pharmaceutical products in materials that create a microbial barrier, preventing contamination from the point of manufacture through to the moment of use. Unlike regular packaging that simply contains a product, sterile packaging must actively maintain sterility over its entire shelf life—guarding against microorganisms, moisture, light, and physical damage.
The Sterile Barrier System (SBS)
The sterile barrier system (SBS) is the minimum packaging required to prevent microbial ingress. According to ISO 11607-1, the SBS must perform three core functions:
- Allow sterilization of the enclosed product
- Provide an acceptable microbial barrier throughout shelf life
- Enable aseptic presentation at the point of use
The barrier must remain intact and uncompromised until the product is deliberately opened by the end user.
Common SBS formats include peel-open pouches, rigid sterilization containers, and sterilization wraps. Each format must be validated to ensure it can withstand the specific sterilization process and maintain integrity throughout storage and handling.
Terminally Sterilized Products
A product is considered terminally sterilized when it is sterilized within its final sterile barrier system, as opposed to being sterilized before packaging. This distinction matters because terminally sterilized products offer greater sterility assurance—the entire sealed package undergoes the sterilization process, eliminating any contamination introduced during packaging assembly.
For medical product businesses, knowing whether a product is terminally sterilized has direct downstream consequences: it shapes validation requirements, labeling decisions, and what regulators expect in your submissions.
Why Sterile Packaging Matters for Medical Products
Patient Safety and Healthcare-Associated Infections
Healthcare-associated infections (HAIs)—including surgical site infections—are among the most preventable yet costly events in healthcare. The CDC reports that SSIs are associated with a 2- to 11-fold increase in mortality risk, with 75% of SSI-associated deaths directly attributable to the infection. Compromised sterile packaging is a direct contributing factor.
An FDA early alert regarding Integra LifeSciences' MediHoney and CVS Wound Gel products highlighted that packaging failures led to a breach in the sterile barrier, resulting in 14 reported serious injuries. Similarly, an FDA recall for Alcon Custom Pak ophthalmic procedure packs was initiated due to incomplete pouch seals, which increased the risk of microbial contamination and subsequent ocular infection.
Product Protection Beyond Sterility
Sterile packaging also protects delicate instruments and devices from physical damage, moisture, and environmental factors during storage, handling, and transport—all of which can affect device functionality before it ever reaches a patient. A punctured pouch or torn seal doesn't just compromise sterility; it can expose sensitive electronics, degrade materials, or allow corrosion.
Business Risk of Non-Compliance
Those same physical failures carry serious downstream consequences for your business. The FDA classifies sterile packaging products such as sterilization pouches as Class II medical devices requiring 510(k) clearance, which means validation is required before use. Non-compliance with ISO 11607, FDA regulations, or GMP standards can result in:
- Invalidated sterile claims and halted distribution
- Costly product recalls and regulatory action
- Reputational damage with healthcare customers
- Legal liability exposure
Types of Sterile Packaging Explained
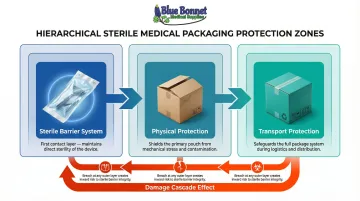
Sterile packaging is organized into three functional layers, each playing a distinct role in maintaining sterility, physical protection, and transport integrity.
Primary Sterile Packaging (The Sterile Barrier System)
Primary packaging is the innermost layer in direct contact with or immediately surrounding the medical device. It forms the actual sterile barrier and must allow sterilant penetration while blocking microbial re-entry.
Common formats include:
- Peel pouches (self-seal and heat-seal)
- Blister packs
- Formed trays with lidding
- Rigid sterilization containers
Primary packaging must be validated to demonstrate that it maintains sterile integrity throughout the product's shelf life and allows for aseptic presentation at the point of use.
Secondary Sterile Packaging (Protective Packaging)
Secondary packaging is the outer protective layer around the primary packaging. It adds physical durability, protects the primary barrier from punctures or tears during handling, and often carries labeling and product information.
Examples include:
- Paperboard cartons
- Outer wraps
- Header bags
Its job is ensuring the primary barrier survives distribution hazards intact — because a compromised secondary layer is often the first step toward a sterility failure.
Tertiary Packaging (Transport Packaging)
Tertiary packaging is used for bulk transport and storage. It protects primary and secondary packaging from mechanical damage and environmental factors like corrosion during shipping.
Examples include:
- Corrugated shipping boxes
- Pallets
- Stretch wrap
How the Three Layers Work Together
These three layers function as a chain. Failure at any layer can compromise the entire sterile chain. For example, a damaged shipping box (tertiary) can puncture the carton (secondary), which in turn can tear the sterile pouch (primary), rendering the device non-sterile.
That failure chain is why packaging selection matters from the start. Match packaging type to the specific device's requirements — size, weight, sensitivity — and the sterilization method being used. Incompatible combinations can cause packaging failure even when each component is individually validated.
Common Formats and Materials Used in Sterile Packaging
Packaging Formats
The most common sterile packaging formats include:
- Peel-open pouches (self-seal and heat-seal): Used for small to medium-sized instruments; allow visual inspection of contents and easy aseptic opening
- Roll stock or reels: Tubing material sealed at both ends; used for custom-sized packages in healthcare facilities
- Rigid sterilization containers: Reusable systems with filters that allow sterilant penetration; used for surgical instrument sets
- Sterilization wraps (woven and nonwoven): Flexible wraps used in healthcare facilities to package instruments for steam sterilization
Format selection drives material selection — and that's where most compliance issues originate.
Packaging Materials
Using the wrong material with the wrong sterilization method leads to packaging failure. All materials must be non-toxic, non-pyrogenic, allow sterilant penetration, and resist microbial ingress after sterilization.
Common materials include:
- Medical-grade paper: Compatible with steam and ethylene oxide (EO) sterilization; commonly used in peel pouches
- Tyvek (spunbonded polyolefin): Compatible with hydrogen peroxide, EO, and radiation sterilization; steam-compatible only under strictly controlled conditions
- Clear plastic films: Used in blister packs and pouches; allow visual inspection
- Paperboard: Used for secondary packaging; provides physical protection
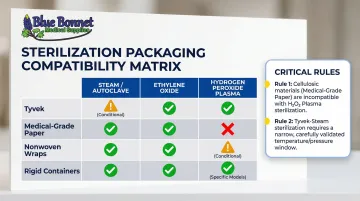
The table below shows which materials are validated for each sterilization method — mismatches here are a common source of regulatory failures.
Material Compatibility by Sterilization Method:
| Material Type | Steam/Autoclave | Ethylene Oxide (EO) | Hydrogen Peroxide (H₂O₂) Plasma |
|---|---|---|---|
| Tyvek (Spunbonded HDPE) | Yes (controlled conditions only) | Yes | Yes |
| Medical-Grade Paper | Yes | Yes | No (neutralizes sterilant) |
| Nonwoven Wraps | Yes | Yes | Varies by manufacturer |
| Rigid Containers | Yes | Yes (via filters) | Varies by manufacturer |
Two compatibility rules require particular attention. First, cellulosic materials — medical-grade paper and linens — are unacceptable for hydrogen peroxide gas plasma sterilization because they neutralize the sterilizing agent. Second, Tyvek with steam is only valid within a narrow window: 121°C to 127°C at 30 psi for 30 minutes, with less than 1.6% shrinkage. Manufacturers must validate each material-method combination before use.
Regulatory Requirements You Need to Know
ISO 11607: The Global Standard
ISO 11607 is the primary global standard for terminally sterilized medical device packaging. It consists of two parts:
- ISO 11607-1: Specifies requirements for materials, sterile barrier systems, and packaging systems
- ISO 11607-2: Specifies validation requirements for forming, sealing, and assembly processes
Compliance with ISO 11607 is expected by regulatory bodies worldwide, including the FDA, which formally recognizes both standards. The standard mandates stability testing to demonstrate that the sterile barrier system maintains integrity over time, with real-time aging studies required to support shelf-life claims.
FDA Oversight and Classification
In the US, sterile packaging products such as sterilization pouches are classified as Class II medical devices under 21 CFR 880.6850 and require 510(k) premarket notification. Recent 510(k) clearances show what FDA expects in practice:
- K153540 (Safe Secure Sterilization Pouch): Validated for gravity/pre-vacuum steam and EO; demonstrated a 5-year pre-sterilization shelf life
- K183356 (Disposable Medical Device Self-seal Sterilization Pouch): Validated for pre-vacuum steam; referenced ISO 11607-1 and ASTM standards for seal integrity
- K241123 (Perpak Sterilization Tyvek Pouch): Validated specifically for hydrogen peroxide sterilization; demonstrated sterility maintenance for 3 years post-sterilization
ANSI/AAMI ST79 Guidelines
ANSI/AAMI ST79 provides guidance specific to sterilization in healthcare facilities, including guidelines on pouch sizing, material selection, and sterilant penetration. This standard is particularly relevant for hospitals and clinical sterile processing environments, where staff package instruments for sterilization on-site.
AAMI ST79 dictates that the shelf life of facility-sterilized items is "event-related" and depends on packaging quality, storage conditions, and handling—not a fixed calendar date.
Sterilization Indicators
Chemical and biological indicators each verify sterilization differently—and both share a key constraint:
- Chemical indicators (CIs): Change color when exposed to a sterilization process; classified into six types by ISO 11140-1 (e.g., Type 1 Process Indicators, Type 5 Integrating Indicators)
- Biological indicators (BIs): Use highly resistant bacterial spores (e.g., Geobacillus stearothermophilus) to confirm microbicidal efficacy
Critical limitation: A "pass" response from a CI or a negative BI indicates exposure to the process but does not prove that the specific item monitored is sterile. Both are used for process validation, not individual item certification.
GMP Compliance
Companies producing or packaging medical products must comply with Good Manufacturing Practice (GMP) standards. GMP governs manufacturing, packaging, storage, and distribution conditions—and non-GMP environments can invalidate sterile claims even when the packaging itself is correct.
Under FDA's Quality Management System Regulation (21 CFR 820.45), manufacturers must maintain documented procedures covering:
- Packaging and labeling controls during production
- Integrity verification during storage and handling
- Traceability through distribution

Key Considerations for Safer Sterile Packaging
The Matching Principle: Validate the Entire System
The packaging material, format, and sterilization method must all be validated together—not assumed to be compatible. Businesses should always verify their packaging is validated for their specific sterilization process and device type, and consult manufacturer Instructions for Use (IFUs).
For example, a Tyvek pouch validated for hydrogen peroxide sterilization may not be validated for steam sterilization, even though Tyvek can technically withstand steam under controlled conditions. Similarly, medical-grade paper validated for steam cannot be used with hydrogen peroxide plasma.
Shelf Life and Storage Conditions
Sterile packaging has a defined shelf life tied directly to storage environment. ISO 11607-1 requires stability testing to confirm the sterile barrier maintains integrity over time, with real-time aging studies initiated within three months of accelerated aging.
Storage conditions matter throughout the product's lifecycle. The following environmental and handling factors can degrade packaging, compromise seals, or introduce micro-perforations that allow microbial ingress:
- Excessive heat or humidity
- Direct light exposure over extended periods
- Rough handling during warehousing or transit
Logistics and Handling Dimension
Sterile packaging integrity can be compromised not only during manufacturing but during warehousing, picking, packing, and shipping. Medical product businesses should work with storage and fulfillment partners who operate under FDA, ISO, and GMP-compliant conditions.
Under FDA regulations, manufacturers retain liability for sterility assurance even when using contract sterilizers or third-party logistics (3PL) providers. Quality agreements must verify that 3PL storage conditions match the exact parameters validated during the manufacturer's ISO 11607-1 stability and performance testing.
That's where choosing the right 3PL partner makes a real difference. Bluebonnet Medical Supplies operates a warehouse designed for sensitive medical items, supporting FDA-compliant storage and ISO and GMP-compliant handling procedures from intake through final shipment.

Frequently Asked Questions
Frequently Asked Questions
What is sterile packaging?
Sterile packaging is specialized packaging designed to create and maintain a microbial barrier around medical devices or pharmaceutical products from the point of manufacture to the point of use, preventing contamination throughout the supply chain.
What are the requirements for sterile packaging?
Requirements include ISO 11607 compliance, FDA clearance, GMP standards, and material compatibility with the chosen sterilization method. Packaging must maintain sterile integrity through shelf life while allowing sterilant penetration and supporting aseptic presentation.
What are the different types of sterile packaging?
The three levels are primary (the direct sterile barrier around the device), secondary (outer protective layer that guards against physical damage), and tertiary (bulk transport packaging).
What should be included in sterile packaging?
Sterile packaging should include the appropriate barrier material, sterilization indicators (chemical or biological), clear labeling with product details and expiration date, and tamper-evident features. Secondary protection is added as needed for distribution.
How does the FDA classify sterile packaging?
In the US, sterile packaging products such as sterilization pouches are classified as Class II medical devices by the FDA and require 510(k) clearance, meaning they must be validated to demonstrate safety and effectiveness before use.
What's the difference between primary and secondary packaging?
Primary packaging is the innermost layer directly surrounding the product and forming the sterile barrier. Secondary packaging is the outer protective layer that guards the primary barrier against physical damage during handling and transport.


