
Introduction
Packaging failure—not device failure—ranks among the leading causes of medical device recalls. A 2025 analysis of FDA recall databases found that seal-integrity failures accounted for 1–4% of all product IDs recalled annually, with 96.7% involving medical devices and 74.8% involving pouches or bags. Each breach in a sterile barrier system directly compromises patient safety by introducing contamination risks, often resulting in high-severity recalls and extensive liability.
Shelf-life validation and testing is the structured process that prevents these failures. This article breaks down what that process involves, which FDA and ISO standards govern it, and how medical device companies can build a compliant, defensible validation program — because a well-engineered device only delivers value if its packaging keeps it sterile through the expiration date.
TLDR
- Shelf-life validation proves packaging maintains sterile integrity from manufacture through expiration
- Process includes IQ/OQ/PQ validation, accelerated and real-time aging, distribution testing, and complete documentation
- ISO 11607 (Parts 1 and 2) and FDA 21 CFR Part 820 govern manufacturer compliance
- Real-time aging must run concurrently with accelerated studies; accelerated data alone does not satisfy regulatory requirements
- Validated packaging integrity requires compliant post-validation storage and distribution to remain effective
What Is Medical Device Packaging Shelf-Life Validation?
Shelf-life validation is the documented process of proving that a medical device's packaging system—specifically its sterile barrier—maintains sterility and physical integrity from manufacture through the claimed expiration date, under real-world handling, storage, and distribution conditions.
Three Validation Types Work Together
Three distinct validation types must all be completed — skipping any one creates regulatory gaps:
- Process validation: Confirms packaging equipment and processes consistently produce acceptable sterile barrier systems
- Shelf-life/stability validation: Uses aging studies to confirm the sterile barrier holds over time
- Distribution/transit validation: Confirms packaging survives the physical stresses of shipping and storage
Key Terminology
ISO 11607-1 defines three core terms you'll encounter throughout validation:
- Sterile barrier system (SBS): The minimum package that prevents microbial ingress and allows aseptic presentation
- Protective packaging: Secondary packaging that shields the SBS from physical damage during distribution
- Packaging system: The complete combination of SBS and protective packaging
Regulatory Framework
Four standards govern shelf-life validation:
- ISO 11607-1:2019: Materials and sterile barrier system requirements
- ISO 11607-2:2019: Forming, sealing, and process validation requirements
- ISO 13485: Quality management system integration
- FDA 21 CFR Part 820: U.S. documentation requirements
The FDA recognizes ISO 11607 compliance as supporting evidence in 510(k) submissions. However, effective December 20, 2026, the FDA will only accept Declarations of Conformity to the 2023 amendments (AMD1:2023), not the base 2019 versions.
Where This Applies
Shelf-life validation applies to any terminally sterilized medical device intended for sterile use at the point of care—pouches, trays, blisters, and wraps. Both primary packaging (the sterile barrier) and secondary/protective packaging must be validated. If your product reaches end users in sterile condition, every layer of packaging in that chain falls within scope.
The Shelf-Life Validation Process: Step by Step
Shelf-life validation isn't a single test but a sequential, multi-phase program. Skipping or rushing any phase exposes gaps that regulatory auditors will flag — and that can force expensive revalidation.
Establish Foundational Requirements First
Before testing begins, confirm three foundations:
- Supplier qualification: Materials must have traceable sourcing and controlled manufacturing
- Material requirements: Packaging materials must be compatible with the sterilization method and provide a microbial barrier before and after sterilization
- Design and performance requirements: The packaging system must enable aseptic delivery and minimize end-user hazards
Phase 1 – Process Validation (IQ/OQ/PQ)
ISO 11607-2 requires a three-step qualification sequence:
Installation Qualification (IQ) confirms equipment is installed correctly and operates as intended.
Operational Qualification (OQ) demonstrates the process produces acceptable results across the limits of the process window, checking seal width, channels, open seals, and aseptic opening capability.
Performance Qualification (PQ) confirms the process is stable and capable under normal manufacturing conditions. ISO 11607-2 explicitly requires samples from three separate production runs to demonstrate reproducibility—not just a one-time result.
Phase 2 – Distribution and Transit Testing
Before aging studies begin, challenge the packaging system against worst-case distribution conditions. Map the real-world distribution channel (truck, air, rail, climate extremes) and select the worst-case product-package combination: the heaviest, most fragile, or highest-puncture-risk configuration.
Applicable standards include:
- ASTM D4169: Standard Practice for Performance Testing of Shipping Containers (FDA-recognized)
- ISTA protocols: 2-Series (Partial Simulation) and 3-Series (General Simulation) for medical devices
Post-transit inspections verify sterile barrier integrity, label legibility, tamper evidence, and aseptic presentation capability.
Phase 3 – Shelf-Life/Aging Studies
Once transit testing confirms the packaging survives distribution, aging studies assess whether it maintains integrity over the full claimed shelf life.
Accelerated Aging
ASTM F1980 uses elevated temperature and humidity in environmental chambers to simulate time passage, allowing manufacturers to project shelf-life performance before real-time data is available. The Arrhenius equation is the basis of accelerated aging calculations—but going too high in temperature can create false positive failures.
Real-Time Aging
Manufacturers must store packaging under ambient conditions for the full claimed shelf life. ISO 11607-1 requires real-time aging to fully validate shelf-life claims. Accelerated aging alone is only acceptable for initial market entry, pending real-time data. Both studies must run concurrently.
Phase 4 – Usability Evaluation and Documentation
ISO 11607:2019 added a usability evaluation requirement. Manufacturers must assess whether clinical end users can aseptically open and dispense the product without contaminating contents.
Three minimum criteria:
- Identifying where to open
- Recognizing the technique required
- Aseptically presenting contents
All phases must be captured in a complete technical file. Required documentation includes:
- Design files and material qualification data
- Validation study protocols and results
- Sterilization validation evidence
- Aging study reports (both accelerated and real-time)
- Labeling controls and failure analysis records
Every document must show clear traceability between the test performed and the regulatory requirement it satisfies.
Key Testing Methods Used in Shelf-Life Validation
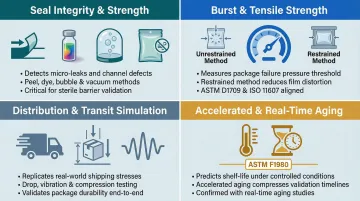
Seal Integrity and Strength Testing
Core methods verify the sterile barrier won't open prematurely or allow microbial ingress:
- Seal strength testing: Peel force measurement per ASTM F88/F88M-23
- Dye penetration testing: Detects channel leaks ≥50 µm per ASTM F1929-23
- Bubble emission testing: Detects gross leaks down to 250 µm per ASTM F2096-11
- Vacuum leak/decay testing: Measures pressure changes indicating seal compromise
Burst and Tensile Strength Testing
Burst strength testing measures the pressure at which packaging fails, confirming it can withstand sterilization and handling stress. Tensile testing evaluates the mechanical strength of the package materials themselves.
Two burst test methods exist:
- ASTM F1140 (Unrestrained): Stress is highest at the middle where the pouch inflates to maximum diameter
- ASTM F2054 (Restrained): Restraining plates distribute stress more uniformly along perimeter seals, providing higher probability of detecting the weakest seal area
Distribution and Transit Simulation Testing
Transit simulation covers the full range of real-world stresses packaging encounters before reaching the end user:
- Vibration (truck, air, and rail transport)
- Compression
- Drop and shock
- Climate and pressure exposure
The sterilization step should be incorporated into the study design where applicable. Testing must use the worst-case product-package combination.
Accelerated Aging Testing (ASTM F1980)
Elevated temperature and humidity conditions compress time. Parameters that must be controlled include temperature, relative humidity, and Q10 factor (typically 2, meaning a 10°C increase doubles the reaction rate).
Inspections at each aging interval verify:
- Sterile barrier integrity
- Label legibility
- Barcode scanning
- Physical/chemical characteristics of materials
Microbial Barrier and Material Property Testing
Aging studies must include evaluation of microbial barrier properties—confirming the packaging prevents microbial ingress through end of shelf life. Physical/chemical material characteristics must also be assessed. Biological safety testing per ISO 10993 may be required depending on device type.
Common Mistakes That Derail Packaging Shelf-Life Validation
Three mistakes account for the majority of failed validations and delayed submissions. Catching them early can save months of rework.
Treating Accelerated Aging as a Substitute for Real-Time Aging
Relying solely on accelerated aging data without initiating concurrent real-time studies is a frequent mistake that can halt regulatory approvals. Manufacturers must run both studies simultaneously — accelerated results support market entry while real-time data confirms validity over the full shelf life.
Skipping or Delaying Material Qualification
Failing to qualify packaging materials for compatibility with the intended sterilization process (ethylene oxide, gamma, or electron beam) upfront leads to failed seal integrity tests, material degradation, and redesign cycles that derail timelines. Material qualification should begin at the earliest design stages, not after prototypes are already in testing.
Inadequate Documentation and Traceability
Incomplete technical files are a leading cause of audit failures and submission rejections. The specific gap regulators flag most often: missing links between test results, risk assessments, and the regulatory requirements they address.
A 2019 FDA Warning Letter to Clinicon Corp cited the firm for failing to validate its packaging process: "You told our investigator that no package integrity testing has been performed and you could not provide evidence to show how the packaging procedure was developed per a protocol... to validate the sealing process."
Build documentation as an auditable, living record throughout the process — not as a retroactive summary assembled before submission.
How Bluebonnet Medical Supplies Supports Your Packaging Compliance
Passing shelf-life validation only protects product sterility if the downstream storage and distribution chain maintains the same controlled conditions under which packaging was validated. Temperature excursions, improper handling, or inadequate storage facilities can compromise validated packaging integrity before a device ever reaches a patient.
Bluebonnet Medical Supplies operates an FDA-cleared, ISO and GMP-compliant warehouse built specifically for medical products. It's the critical link between successful shelf-life validation and compliant, intact delivery.
Compliance-Focused Warehousing and Distribution
Bluebonnet's capabilities directly support packaging compliance:
- Proper storage of sensitive medical items in a warehouse designed for medical products, not general consumer goods
- FDA-compliant packing and shipping procedures that maintain sterile barrier integrity during outbound distribution
- HIPAA-safe handling for products requiring protected health information safeguards
- Returns processing with product restoration that includes inspection and testing to ensure returned items meet usability and safety standards

Personalized Approach to Medical Device Logistics
As a boutique 3PL, Bluebonnet builds logistics solutions around each client's specific regulatory requirements—device class, destination market, storage conditions, and documentation needs. Bluebonnet handles the distribution chain so medical device businesses can focus on product development and growth.
That personalized model translates into practical compliance support:
- Tailored storage protocols matched to each product's validated environmental conditions
- Destination-specific shipping procedures for domestic and international orders, including customs documentation
- Dedicated account handling so regulatory questions reach someone who knows your product—not a general support queue
Frequently Asked Questions
What is the process of medical device packaging validation?
Validation covers four key phases: foundational requirements (material and supplier qualification), process validation (IQ/OQ/PQ per ISO 11607-2), distribution/transit testing, and shelf life aging studies (accelerated plus real-time). Each phase must be documented in a Device History Record or Technical File per ISO 13485.
What are the main types of validation for medical device packaging?
ISO 11607 requires three validation types: process validation (IQ/OQ/PQ), shelf life and stability validation (accelerated and real-time aging), and distribution/transit validation. A fully compliant packaging system must satisfy all three.
Why are three batches required for medical device packaging validation?
Performance Qualification (PQ) under ISO 11607-2 requires samples from three separate production runs to demonstrate that the packaging process is consistently capable under normal manufacturing conditions, not just a one-time result.
What are the key packaging requirements for medical devices?
Key requirements cover sterile barrier integrity throughout the product's shelf life, compatibility with the sterilization method, adequate protection during distribution, aseptic presentation capability, and full documentation meeting ISO 11607 and FDA 21 CFR Part 820.
What is the ISO standard for medical device packaging validation?
ISO 11607 is the primary standard. ISO 11607-1 covers material and sterile barrier system requirements, while ISO 11607-2:2019 covers process validation requirements. ISO 13485 and FDA 21 CFR Part 820 also apply.
What is the difference between accelerated aging and real-time aging for medical device packaging?
Accelerated aging uses elevated temperature and humidity per ASTM F1980 to simulate shelf life in a compressed timeframe, enabling faster market launch. Real-time aging stores packaging under standard conditions for the full claimed shelf life. Both are required; accelerated aging alone is not an accepted substitute under ISO 11607.


