
Introduction
A medical device that reaches a patient with compromised packaging can cause contamination, serious injury, or death. In 2021, Exactech recalled thousands of joint implants after packaging defects missing an oxygen barrier layer led to 2,600 lawsuits and pushed the company into bankruptcy by 2024.
Shelf-life validation is the documented, systematic process that proves packaging maintains sterile barrier performance from manufacturing through a device's entire claimed shelf life. It's not a regulatory formality — it's what separates compliant products from recalled ones.
For medical device manufacturers, this process confirms that packaging protects devices against microbial contamination and physical damage under real-world storage conditions. Skipping it carries steep costs: failed FDA submissions, product recalls, customs rejections, and liability exposure. This guide covers the key testing standards, validation methods, and compliance steps you need to get it right.
TL;DR
- Shelf-life validation proves medical device packaging maintains sterile barrier integrity for the full labeled shelf life
- The process combines accelerated aging (ASTM F1980) for rapid data with real-time aging confirmation
- Testing includes seal strength, burst, dye penetration, bubble emission, and distribution simulation
- Regulatory compliance requires documented validation per ISO 11607, FDA 21 CFR 820.75, and EU MDR prior to market entry
- Accelerated aging results are provisional; real-time aging data is required to finalize validation
What Is Medical Device Packaging Shelf-Life Validation?
Shelf-life validation is the documented process of proving that a packaging system maintains its sterile barrier properties—blocking microbial contamination and physical damage—throughout a device's stated shelf life under specified storage conditions.
Understanding the Sterile Barrier System
The sterile barrier system (SBS) is the inner packaging that directly prevents contamination from reaching the medical device. The protective packaging consists of outer layers that absorb distribution stress during shipping and handling. Both layers require validation, though shelf-life studies center on proving the sterile barrier holds up over time.
Validation vs. Shelf-Life Validation
The two are related but distinct:
- Packaging validation — confirms the system design works at the outset
- Shelf-life validation — confirms it continues to work over time
A packaging system must pass design validation before shelf-life claims can be established.
Business Consequences of Inadequate Validation
Failing to validate shelf-life creates serious business risks:
- FDA submission rejections due to incomplete or insufficient validation data
- Product recalls when packaging fails in the field
- Customs rejections for international shipments lacking proper documentation
- Liability exposure from patient safety incidents linked to packaging failures
According to an FDA recall analysis from 2011-2013, packaging-related failures accounted for 60 documented recalls, with the majority described as "loss of integrity" (24 cases), "defect seals" (15 cases), and "puncture" (12 cases). More recent metadata analysis of FDA recalls confirms that seal-related failures continue to drive recalls, almost exclusively in medical device packaging such as trays and pouches.
Regulatory Standards That Govern Packaging Shelf-Life Testing
ISO 11607: The Global Standard for Sterile Medical Device Packaging
ISO 11607 is the primary global standard for sterile medical device packaging. It consists of two parts:
- Part 1 covers materials, sterile barrier systems, and packaging systems
- Part 2 covers validation of forming, sealing, and assembly processes
Compliance with ISO 11607 is required for FDA submissions and CE marking under EU MDR. As of March 2024, the European Commission officially harmonized the amended ISO 11607-1 and -2 (2020/A1:2023) under EU MDR via Implementing Decision (EU) 2024/815 — granting presumption of conformity for CE marking.
ASTM F1980: Accelerated Aging of Sterile Barrier Systems
ASTM F1980 is the key standard for accelerated aging of sterile barrier systems. It's based on the Q10 principle (van't Hoff equation): chemical reaction rates approximately double for every 10°C increase in temperature, allowing manufacturers to use elevated temperature and humidity to simulate years of aging in weeks.
Critical limitation: ASTM F1980 explicitly states that accelerated aging data is only sufficient until real-time data is available.
Regulators expect both datasets: accelerated aging supports initial market launch, while real-time aging confirms the claimed shelf life over the full product lifecycle.
Supporting ASTM and ISTA Standards
Beyond ISO 11607 and ASTM F1980, a set of supporting standards covers specific test methods — each addressing a distinct failure mode or distribution risk. The FDA recognizes these consensus standards for compliance submissions:
| Standard | Purpose | Recognition Status |
|---|---|---|
| ASTM F88 | Peel/seal strength testing | FDA Recognized |
| ASTM F1929 | Dye penetration for detecting seal leaks ≥50 µm | FDA Recognized |
| ASTM F2096 | Bubble emission for detecting gross leaks ≥250 µm | FDA Recognized |
| ASTM F1140 | Burst testing for internal pressurization resistance | FDA Recognized |
| ASTM F2054 | Burst testing of flexible package seals | FDA Recognized |
| ASTM D4169 | Distribution simulation testing | FDA Recognized |
| ISTA Protocols | Alternative distribution simulation methods | Industry standard |

How Shelf-Life Validation Works – Step by Step
Shelf-life validation follows a defined sequence where each stage builds on the last. Miss a step or run them out of order and you risk study restarts, regulatory rejections, or failed field performance.
Step 1 – Define the Shelf-Life Claim and Packaging Requirements
Identify the target shelf life (e.g., 2, 3, or 5 years) based on clinical need and distribution realities. This number drives everything: aging study duration, test interval selection, and documentation scope.
Required inputs:
- Device type and classification
- Sterilization method (EtO, gamma, e-beam, steam)
- Storage conditions (temperature and humidity)
- Intended distribution geography
Step 2 – Select and Qualify Packaging Materials
Material qualification must happen before aging studies begin. Materials must be compatible with the chosen sterilization method:
- Gamma/e-beam radiation causes polymer chain scission and embrittlement, degrading some polymers
- EtO sterilization requires gas-permeable materials like Tyvek with high moisture vapor transmission rates for proper aeration
- Steam sterilization demands materials that withstand temperatures up to 121-127°C and pressure differentials
Incompatible materials will fail during aging studies — triggering re-qualification, re-testing, and study timelines that can push launch dates back by 3-6 months.
Step 3 – Run Accelerated Aging Studies
Accelerated aging uses elevated temperature and humidity (per ASTM F1980) to simulate the passage of time in a compressed study window. Manufacturers run accelerated aging first to get preliminary shelf-life data fast while real-time aging studies run in parallel.
Using the Q10 factor of 2.0, typical compressed timeframes are approximately:
- 1-year claim: ~46 days
- 2-year claim: ~92 days
- 3-year claim: ~137 days
- 5-year claim: ~229 days
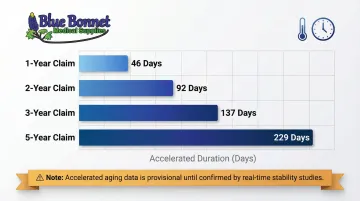
Common mistake: Treating accelerated aging results as final validation without completing real-time confirmation. The FDA explicitly requires that real-time aging studies run in parallel to confirm accelerated predictions.
Step 4 – Conduct Integrity and Performance Testing at Each Interval
Test samples are pulled at defined intervals (e.g., T=0, T=6 months, T=12 months, end of life) and subjected to seal integrity, distribution simulation, and sterile barrier testing. Results at each interval must meet pre-established acceptance criteria—not just pass at the end point.
Storage conditions during the aging study must be tightly controlled. That control doesn't end when validation does — packaging stored at the wrong temperature, humidity, or handling environment after validation can fail in the field despite a clean study result. A device that passed every test on paper can still breach sterile barrier if warehouse conditions aren't maintained to the same standards used during testing.
Step 5 – Document Results and Maintain Validation Records
Every step must be captured in a Packaging Validation Report (PVR) that includes:
- Test protocols with pre-approved acceptance criteria
- Sample sizes and sampling plans
- Raw data and test results
- Deviations and corrective actions
- Conclusions and shelf-life determination
This documentation is what regulators review during submissions and audits. Incomplete or inconsistent records are a leading cause of FDA warning letters and submission rejections.
Core Testing Methods Used in Packaging Shelf-Life Validation
Shelf-life validation draws on several complementary test methods, each revealing different failure modes. A complete validation protocol uses multiple methods to confirm the sterile barrier holds under every realistic stress condition — seal integrity, distribution forces, and long-term aging each require their own approach.
Seal Integrity Tests
These methods target the seal itself — detecting defects, measuring bond strength, and confirming barrier integrity across a sample population.
- Visual Inspection (ASTM F1886): Detects gross defects down to 75 µm in flexible packaging. First line of defense for obvious seal problems.
- Peel/Seal Strength (ASTM F88): Tracks whether bond strength degrades during aging, which compromises sterile barrier integrity if left unchecked.
- Dye Penetration (ASTM F1929): Detects channel leaks ≥50 µm in porous packaging like Tyvek pouches. Destructive; used on sample populations.
- Bubble Emission (ASTM F2096): Identifies breaches ≥250 µm in Tyvek pouches and thermoformed trays via internal pressurization. Also destructive.
- Burst/Creep Testing (ASTM F1140/F2054): Confirms packaging withstands pressure changes during distribution, including air transport.
For higher-volume validation programs, vacuum decay (ASTM F2338) and airborne ultrasound offer non-destructive alternatives that provide deterministic, quantitative results without consuming sample units.
Distribution Simulation Testing
Seal integrity tests confirm the barrier exists — distribution simulation tests whether packaging keeps that barrier intact through the entire shipping journey. ASTM D4169 and ISTA protocols replicate the physical stresses devices face in transit:
- Vibration during ground and air transport
- Drops from handling
- Stacking and compression loads
- Pressure changes during air transport
- Temperature and humidity swings
Manufacturers run distribution simulation at T=0 (to validate initial design) and at the end of claimed shelf life (to confirm aged packaging survives the shipping journey intact). ISO 11607 requires testing on the "worst-case" packaged configuration — the heaviest device, sharpest edges, or most challenging contents.
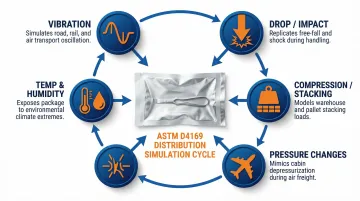
For medical devices, manufacturers typically use ASTM D4169 Distribution Cycle 13 (DC-13) at Assurance Level I or II, which simulates intercity air freight and local motor freight. ISTA 3A is a widely accepted alternative offering rigorous simulation of parcel delivery conditions.
Accelerated vs. Real-Time Aging: Understanding Both
Accelerated aging (ASTM F1980) generates rapid data to support initial launch decisions. It uses elevated temperature and humidity to compress years into weeks.
Real-time aging runs simultaneously under ambient conditions to provide long-term confirmation. When real-time data diverges from accelerated predictions by more than acceptable tolerances, you must revise the shelf-life claim — the accelerated data does not override it.
Critical regulatory expectation: FDA guidance and ISO 11607-1 explicitly state that accelerated aging is only provisional until real-time data is available. You must initiate real-time studies before commercialization.
How Bluebonnet Medical Supplies Supports Your Packaging Compliance
Shelf-life validation only holds up if the supply chain respects the same conditions the lab tested for. Bluebonnet Medical Supplies is an ISO and GMP-compliant 3PL partner with FDA-compliant medical packaging services — built specifically to maintain those conditions through every stage of storage, handling, and distribution.
Warehouse Built for Sensitive Medical Items
Bluebonnet's warehouse is designed specifically for sensitive medical devices — with environmental controls and handling protocols aligned to FDA, ISO, GMP, and HIPAA-safe standards. Storage conditions in the supply chain match the conditions your packaging was validated under.
Compliance Throughout the Supply Chain
Bluebonnet integrates regulatory compliance directly into daily warehouse and fulfillment operations:
- Maintains controlled environmental conditions suited to sensitive medical devices
- Applies FDA-compliant labeling at the point of packaging and fulfillment
- Handles sterile packaging to prevent physical compromise during pick-and-pack
- Documents inventory with audit-ready tracking standards
- Screens international shipments for compliance to prevent customs delays

Supporting Multi-Channel Distribution
Whether you're selling through Amazon, your own website, or shipping internationally, Bluebonnet removes compliance friction for medical device companies — so packaging arrives at healthcare facilities in the condition your shelf-life validation was designed to guarantee.
Beyond distribution, Bluebonnet also supports the full product lifecycle:
- Inspects and restores returned items through quality assurance testing
- Provides custom packaging and labeling with FDA-compliant medical packaging solutions
- Offers sterile packaging options for medical devices
- Performs kitting and assembly in a controlled environment
For detailed information about how Bluebonnet's environmental controls, handling protocols, and documentation systems can support your specific packaging compliance requirements, contact us at info@bbmstx.com.
Frequently Asked Questions
What types of tests are performed in medical device package testing?
Medical device package testing covers three main categories, with specific tests selected based on package format, materials, and sterilization method:
- Seal integrity: Visual inspection, peel strength (ASTM F88), dye penetration (ASTM F1929), burst testing, bubble emission (ASTM F2096)
- Distribution simulation: ASTM D4169 or ISTA protocols
- Aging studies: Accelerated (ASTM F1980) and real-time
What standards apply to medical device package testing?
ISO 11607 is the primary global standard for sterile medical device packaging, supported by ASTM standards including F1980 (accelerated aging), F88 (seal strength), F1929 (dye penetration), F2096 (bubble emission), and D4169 (distribution simulation). The FDA and EU MDR build additional requirements on top of these consensus standards.
Which tests are required for all types of medical device packages?
ISO 11607 universally expects visual inspection, seal strength testing, and sterile barrier integrity testing across all package types. Distribution simulation and aging studies—both accelerated and real-time—are also required to establish and confirm shelf-life claims. Exact requirements vary by device class and sterilization method.
What is accelerated aging in medical device packaging?
Accelerated aging uses elevated temperature and humidity per ASTM F1980 to simulate years of shelf life in weeks or months, based on the Q10 principle that reaction rates double for every 10°C increase. It establishes provisional shelf-life claims alongside real-time aging studies, which provide the final confirmation.
How long does shelf-life validation testing take?
Accelerated aging studies can compress multi-year claims into weeks or months (e.g., ~92 days for a 2-year claim at 55°C). However, complete validation including real-time confirmation, testing at all intervals, documentation review, and regulatory submission typically takes several months to over a year depending on the claimed shelf life.
What happens if medical device packaging fails shelf-life testing?
Failure triggers a root cause investigation into sealing parameters, material compatibility, or storage conditions, followed by corrections and re-validation—a process that can delay product launch by months. Failures that go undetected reach the field as product recalls, FDA warning letters, and patient safety incidents, as the Exactech bankruptcy case illustrates.


