
This guide breaks down what you need to know: the FDA's official definition of a medical device, how devices are classified by risk level, the main pathways to market clearance or approval, and what compliance means after your device is authorized. Whether you manufacture, distribute, or warehouse medical products, understanding these fundamentals protects your business from costly enforcement actions.
TLDR
- The FDA classifies devices into three risk-based classes (I, II, III) that determine which regulatory pathway and controls apply
- Most devices reach market through 510(k) clearance (substantial equivalence) or PMA approval (rigorous clinical review)
- Post-market obligations, including adverse event reporting and quality system compliance, continue throughout a device's lifecycle
- FDA registration of your facility is separate from product clearance or approval
- Distributors and 3PLs must maintain FDA-compliant handling, storage, and labeling throughout the supply chain
What Is a Medical Device? The FDA's Official Definition
The Statutory Baseline
The FDA defines a medical device under Section 201(h)(1) of the Food, Drug, and Cosmetic Act (FD&C Act) as:
"An instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article... intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease... and which does not achieve its primary intended purposes through chemical action within or on the body."
This definition has one critical distinguishing feature: devices do not work through chemical action in the body, which is what separates them from drugs. The FDA's Center for Devices and Radiological Health (CDRH) governs all firms that manufacture, repackage, relabel, or import medical devices sold in the United States.
Intended Use Determines Classification
Intended use is the decisive factor the FDA evaluates when classifying a product. Under 21 CFR 801.4, intended use refers to the objective intent shown through labeling claims, advertising, or written statements. The same physical product can be classified differently depending on how it's marketed:
A wearable sensor marketed for general wellness tracking = likely not a medical device
The same sensor marketed to diagnose arrhythmias = Class II medical device requiring 510(k) clearance
A blood pressure monitor marketed as a fitness tool = consumer electronics; marketed to manage hypertension = regulated medical device
Moving from a general indication to a specific medical indication can alter your regulatory standing entirely and trigger premarket review requirements.
Medical Devices Extend Beyond Surgical Tools
The statutory definition covers far more than scalpels and stethoscopes:
- Software as a Medical Device (SaMD): Software intended for medical purposes that performs these purposes without being part of a hardware device
- In Vitro Diagnostics (IVDs): Reagents, instruments, and systems used to diagnose disease through examination of specimens taken from the human body
- Implants: Pacemakers, joint replacements, cochlear implants
Before pursuing any regulatory pathway, verify your product's classification using the FDA Product Classification Database. Getting this wrong early can mean costly pathway changes later — whether you're a manufacturer, importer, or distributor handling these products.
The Three FDA Medical Device Classes Explained
Risk-Based Classification Framework
The FDA categorizes all medical devices into three classes based on risk level and the degree of regulatory control necessary to ensure safety and effectiveness. Classification determines which controls and regulatory pathways apply to your product.
The FDA has established classifications for approximately 1,700 different generic device types, grouped into 16 medical specialty panels.
Class I: Low Risk, General Controls
Class I devices present minimal potential for harm and are subject only to General Controls:
- Establishment registration and device listing
- Proper labeling and instructions for use
- Good Manufacturing Practice (GMP) requirements
- Prohibition against adulteration and misbranding
Examples: Bandages, tongue depressors, hospital beds, elastic bandages
Regulatory pathway: Approximately 74% of Class I devices are exempt from premarket notification (510(k)), though they must still comply with General Controls.
Class II: Moderate Risk, Special Controls
Class II devices pose moderate risk, requiring both General Controls and Special Controls because general controls alone are insufficient. Special Controls include:
- Performance standards
- Post-market surveillance requirements
- Specific labeling requirements
- Patient registries
- FDA guidance documents
Examples: CT scanners, infusion pumps, surgical gloves, pregnancy test kits
Regulatory pathway: Most Class II devices require 510(k) clearance before marketing.
Class III: High Risk, Premarket Approval
Class III devices support or sustain human life, are substantially important in preventing impairment of health, or present potential unreasonable risk of illness or injury. These require the most rigorous review.
Examples: Implantable pacemakers, cochlear implants, replacement heart valves, deep brain stimulators
Regulatory pathway: Premarket Approval (PMA) is required, demanding extensive clinical evidence of safety and effectiveness.
Quick Reference Comparison
| Class | Risk Level | Controls | Typical Pathway | Example |
|---|---|---|---|---|
| I | Low | General Controls only | 510(k)-exempt (74%) | Bandages |
| II | Moderate | General + Special Controls | 510(k) clearance | Infusion pumps |
| III | High | General Controls + PMA | PMA approval | Pacemakers |

Regulatory Pathways to Market: 510(k), PMA, and Exemptions
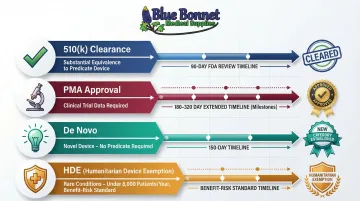
510(k) Clearance: Proving Substantial Equivalence
The 510(k) pathway requires demonstrating that your device is "substantially equivalent" to a legally marketed predicate device. Your device is substantially equivalent if it:
- Has the same intended use as the predicate and the same technological characteristics, or
- Has the same intended use and different technological characteristics that don't raise new safety/effectiveness questions, with submitted data demonstrating equivalent safety and effectiveness
Critical distinction: A successful 510(k) results in clearance, not approval. The FDA issues an order clearing the device for commercial distribution.
Timeline: Under MDUFA V performance goals, the FDA aims to decide 95% of 510(k) submissions within 90 FDA Days.
Premarket Approval (PMA): Highest-Burden Pathway
PMA is the most stringent device marketing application required under FDA regulations (21 CFR Part 814). The FDA approves a PMA only when the application provides sufficient valid scientific evidence that the device is safe and effective for its intended use.
What's required:
- Extensive clinical trial data
- Manufacturing and quality system documentation
- Proposed labeling
- Risk analysis
Timeline: 180 FDA days for decisions without advisory committee review; 320 FDA days with advisory committee input.
Unlike 510(k) clearance, PMA results in formal FDA approval — a meaningful legal distinction that affects post-market obligations and labeling requirements.
The De Novo Pathway: Opening New Categories
The De Novo pathway addresses novel devices with no legally marketed predicate, where general controls alone or general and special controls provide reasonable assurance of safety and effectiveness.
A granted De Novo establishes a new classification regulation, meaning your device becomes a predicate for future 510(k) submissions. In 2025, CDRH authorized 124 novel medical devices, many through De Novo.
Timeline: 150 FDA days under MDUFA V goals.
510(k) Exemptions
Many Class I and some Class II devices are exempt from premarket submission but must still comply with General Controls. "Exempt" does not mean "unregulated." You must still:
- Register your establishment
- List your device
- Follow GMP requirements
- Maintain proper labeling
Humanitarian Device Exemption (HDE)
The HDE pathway encourages development of devices for rare conditions affecting fewer than 8,000 individuals in the U.S. per year. HDEs are exempt from effectiveness requirements but must demonstrate that probable benefit outweighs risk of injury.
Post-Market Obligations: What Happens After Your Device Is Cleared
Clearing the FDA is a milestone — not a finish line. Once a device is on the market, manufacturers and distributors must meet ongoing compliance obligations that span reporting, quality systems, surveillance, and labeling.
Medical Device Reporting (MDR)
Under 21 CFR Part 803, manufacturers must report specific events to the FDA:
| Entity | Event Type | Reporting Deadline |
|---|---|---|
| Manufacturer | Death, serious injury, malfunction | 30 calendar days |
| Manufacturer | Unreasonable risk of substantial harm | 5 work days |
| Importer | Death, serious injury | 30 days (to FDA & manufacturer) |
| User Facility | Death | 10 work days (to FDA & manufacturer) |
Failure to maintain proper MDR systems is a leading cause of FDA enforcement. In fiscal year 2025, 38 of 54 device Warning Letters concerned Quality System Regulations, with CAPA and complaint handling failures dominating.

Quality Management System Regulation (QMSR)
Effective February 2, 2026, the legacy Quality System Regulation becomes the Quality Management System Regulation (QMSR), incorporating ISO 13485:2016 by reference while retaining specific FDA requirements.
The QMSR covers:
- Design controls
- Production and process controls
- Record-keeping and documentation
- Complaint handling procedures
- Corrective and Preventive Actions (CAPA)
ISO 13485 certification does not replace FDA inspections or premarket requirements — the FDA will not accept ISO certificates as a substitute for its own regulatory oversight.
Section 522 Post-Market Surveillance Studies
The FDA can require manufacturers to conduct post-market surveillance studies for certain Class II and Class III devices already on the market. Order volume has remained relatively low but consistent in recent years:
- FY2021: 2 orders
- FY2022: 1 order
- FY2023: 5 orders
- FY2024: 2 orders
- FY2025: 2 orders
Labeling Compliance
Device labeling must stay current for as long as the product is on the market. Updates may be required when:
- Post-market surveillance uncovers new safety signals
- Adverse event reports indicate a labeling gap
- New clinical data changes the risk-benefit profile
- Regulatory guidance shifts requirements
Labeling failures are often discovered during FDA inspections, making routine label reviews a practical compliance checkpoint.
FDA Registration vs. FDA Approval: Key Differences
Registration Is Not the Same as Approval
FDA registration means your facility is listed with the FDA. FDA clearance or approval means the FDA has reviewed and authorized your specific device for marketing. These are separate requirements that are frequently confused — and that confusion can trigger enforcement action, including Warning Letters and misbranding violations.
Establishment Registration and Device Listing
Under 21 CFR Part 807, owners or operators of establishments involved in producing and distributing medical devices must:
- Register annually with the FDA
- List their devices electronically
- Update registration and listings between October 1 and December 31 each year
Registration alone does not mean any of the following:
- Your device has been reviewed or cleared
- The FDA endorses your product
- You have met premarket requirements for marketing
The Marketing Misrepresentation Trap
Because registration and approval are distinct, federal regulations specifically address the gap. Under 21 CFR 807.39, representations that create an impression of official approval based solely on registration are prohibited. The FDA does not issue device registration certificates, and any claim suggesting otherwise constitutes misbranding.
Real enforcement examples:
- Fast Masks USA LLC received a Warning Letter for selling unauthorized COVID-19 tests while falsely claiming products were "FDA Approved & Authorized"
- Professional World Store was cited for displaying a fake "Certificate of Conformity" with an unauthorized FDA logo
How to Verify a Device's Status
Use these public databases at accessdata.fda.gov:
- 510(k) Premarket Notification database for cleared devices
- PMA database for approved devices
- Product Classification database for device classifications
- Establishment Registration & Device Listing database for facility registrations
How FDA Compliance Affects Your Medical Device Supply Chain
The $16.5 Billion 3PL Market
The complexity of medical device distribution is fueling rapid growth in specialized logistics. The U.S. medical device 3PL market was valued at $10.14 billion in 2024 and is projected to reach $16.58 billion by 2030, rising at 8.5% CAGR. This growth reflects the need for temperature regulation, precision handling, and strict regulatory traceability.
Distributor and 3PL Regulatory Responsibilities
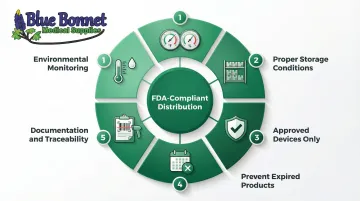
FDA compliance extends beyond manufacturers. Under 21 CFR 820.140-160, distributors and third-party logistics providers must establish procedures for handling, storage, and distribution to prevent mix-ups, damage, deterioration, contamination, or other adverse effects.
Required controls include:
- Environmental monitoring (temperature, humidity)
- Proper storage conditions for sensitive devices
- Ensuring only approved devices are distributed
- Preventing distribution of expired or deteriorated devices
- Maintaining proper documentation and traceability
Non-compliant handling can void a device's regulatory status and trigger enforcement action. A 2026 Warning Letter to Diasol, Inc. illustrates how seriously the FDA takes storage conditions—the agency cited salt erosion damaging the concrete substrate and door breaches compromising temperature, humidity, and contamination control. These failures are the same risks that make choosing a compliant distribution partner a regulatory decision, not just a logistics one.
International Shipping Compliance
Devices exported from or imported to the U.S. must meet FDA labeling and documentation standards to clear customs. Common import refusal charges include:
- No 510(k) clearance on file for devices introduced after 1976
- Establishment not registered under Section 510 of the FD&C Act
- Missing or non-English labeling, which is required under 21 CFR 801.15
Working with Compliant Distribution Partners
For medical device companies shipping domestically or internationally, your 3PL partner's compliance posture is part of your own regulatory footprint. Bluebonnet Medical Supplies provides warehousing, fulfillment, and shipping services built around FDA, ISO, and GMP requirements—covering environmental controls, compliant labeling, and documentation procedures from storage through final delivery. That means fewer customs holds, a cleaner audit trail, and one less compliance gap to manage.
Frequently Asked Questions
What is the official definition of a medical device?
Under Section 201(h) of the FD&C Act, a medical device is any instrument, apparatus, implant, or similar article intended for diagnosing, curing, mitigating, treating, or preventing disease. The key distinction from drugs: a device's primary purpose is not achieved through chemical action in the body.
What is the FDA classification of medical devices?
The FDA classifies all medical devices into Class I (low risk), Class II (moderate risk), or Class III (high risk) based on the level of regulatory control necessary to ensure safety and effectiveness. Classification determines which premarket pathway and ongoing controls apply to each device.
What is a class 1 medical device FDA requirement?
Class I devices must comply with General Controls—including establishment registration, device listing, proper labeling, and adherence to Good Manufacturing Practices. Most Class I devices (approximately 74%) are exempt from premarket review but remain subject to these baseline regulatory requirements.
What does FDA registered medical device mean?
FDA registration means the manufacturer's facility is listed with the FDA — not that the device has been reviewed or approved. Registration and market authorization (clearance or approval) are separate requirements. The FDA does not issue registration certificates, and claiming that registration implies FDA approval constitutes misbranding.
How to check if a medical device is FDA approved?
Use the FDA's public databases at accessdata.fda.gov: search the 510(k) Premarket Notification database for cleared devices and the PMA database for approved devices. You can also verify establishment registration and device listings through the Registration & Listing database.
What is the difference between ISO 13485 and the FDA?
ISO 13485 is an international quality management system standard for medical device manufacturers, while FDA regulation is a U.S. government legal framework. ISO 13485 compliance supports FDA requirements but does not replace them — ISO certification does not substitute for FDA inspections or premarket submissions. The February 2026 QMSR incorporates ISO 13485 by reference as part of this alignment.


