
ISO standards serve as the global benchmark for medical device quality, safety, and regulatory alignment. Whether you're manufacturing Class II diagnostic equipment, distributing sterile surgical instruments, or exporting devices to the EU and Canada, understanding the critical ISO standards—and how they intersect with FDA regulations—is no longer optional. With the FDA's Quality Management System Regulation (QMSR) deadline approaching in February 2026, manufacturers and their supply chain partners must act now to align their operations with ISO 13485:2016 and related technical standards.
TLDR: Key Takeaways
- The FDA's QMSR legally incorporates ISO 13485:2016 by February 2, 2026—making alignment mandatory for U.S. market access
- ISO 13485 covers quality management, but separate standards for risk, biocompatibility, software, and sterilization also apply to your device
- FDA recognition of consensus standards like ISO 14971 and IEC 62304 can streamline submissions, but never substitutes for legal clearance
- Warehouses and logistics partners must maintain ISO and GMP compliance to preserve device conformity throughout distribution
What Are ISO Standards for Medical Devices?
The International Organization for Standardization (ISO) is an independent, non-governmental body that develops consensus-based standards through collaboration among global experts, industry representatives, and regulatory authorities. For medical devices, these standards define requirements for quality, safety, performance, and regulatory compliance throughout the entire product lifecycle—from initial design through manufacturing, distribution, and post-market surveillance.
Voluntary but Practically Mandatory
ISO standards are technically voluntary and don't carry the force of law on their own. However, regulatory bodies worldwide—including the FDA, the European Commission under the Medical Device Regulation (MDR), and Health Canada—formally recognize or harmonize specific ISO standards. This recognition transforms them into practical requirements for market access.
The FDA's upcoming Quality Management System Regulation (QMSR) makes this concrete: it legally incorporates ISO 13485:2016, making conformance mandatory for U.S. distribution by February 2026.
Normative vs. Informative Elements
When applying ISO standards, manufacturers must distinguish between two types of content:
- Normative elements set mandatory "shall" requirements that must be met for compliance and certification
- Informative elements provide guidance, examples, and context to help interpret requirements but cannot be used to claim conformance
Missing this distinction is one of the most common reasons manufacturers face audit findings—requirements get treated as guidance, or guidance gets over-applied as a binding rule.
ISO 13485: The Foundation of Medical Device Quality Management
ISO 13485 is the internationally recognized standard for Quality Management Systems (QMS) designed specifically for medical device manufacturers and their supply chains. It specifies the organizational processes, documentation, and controls needed to consistently produce safe, effective medical devices that meet both regulatory and customer requirements.
What ISO 13485 Certification Involves
Achieving ISO 13485 certification requires a formal third-party audit conducted by an accredited certification body. The process evaluates your documented QMS across critical areas:
- Design controls and development processes
- Production and process validation
- Complaint handling and adverse event reporting
- Corrective and preventive actions (CAPA)
- Regulatory reporting and documentation
- Supplier management and purchasing controls
Certification requires ongoing maintenance. Organizations undergo regular surveillance audits, typically conducted annually, with full recertification every three years.
ISO 13485 vs. ISO 9001: Key Differences
The two standards share a QMS foundation but are built for different goals:
| Aspect | ISO 9001 | ISO 13485 |
|---|---|---|
| Industry scope | General-purpose, any industry | Medical device-specific |
| Primary focus | Customer satisfaction, continual improvement | Regulatory compliance, risk management |
| Performance emphasis | Business growth metrics | Safety and effectiveness |
| Flexibility | Allows exclusions based on applicability | Stricter requirements, fewer exclusions |

Many device companies maintain both certifications: ISO 9001 for overall business quality and ISO 13485 for regulatory compliance.
Supply Chain Implications
ISO 13485 reaches well beyond finished device manufacturers. The standard applies to any organization involved in the product lifecycle, including:
- Sub-tier component suppliers
- Contract manufacturers
- Sterilization service providers
- Logistics and fulfillment partners
Medical device companies increasingly require their entire supply chain to hold or align with ISO 13485, ensuring product conformity from raw materials through final delivery.
FDA Compliance Reality
ISO 13485 provides a strong framework that aligns closely with FDA Quality System Regulation requirements under 21 CFR Part 820 — but the two are not interchangeable. The FDA will not accept ISO certificates as a substitute for inspections or statutory obligations. That said, companies certified under ISO 13485 are better positioned to meet FDA requirements and simplify compliance across multiple international markets at once.
Beyond ISO 13485: Other Key ISO Standards Medical Device Companies Must Know
ISO 13485 establishes your QMS framework, but multiple additional standards govern specific technical areas. Which standards apply to your device depends on its type, intended use, and the markets you're entering — so knowing the full landscape matters.
ISO 14971: Risk Management for Medical Devices
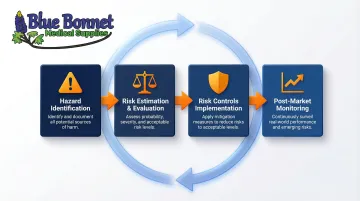
ISO 14971:2019 is the foundational risk management standard for medical devices. It specifies a systematic process for:
- Identifying hazards throughout the device lifecycle
- Estimating and evaluating risks using objective criteria
- Implementing risk controls (design changes, protective measures, information for safety)
- Monitoring risk management effectiveness post-market
The FDA fully recognizes ISO 14971:2019 (Recognition #5-125), making it a standard expectation in 510(k) submissions, PMA applications, and design control documentation. In the EU, the harmonized version (EN ISO 14971:2019) is critical for demonstrating MDR compliance in technical files.
ISO 10993: Biological Evaluation of Medical Devices
The ISO 10993 family comprises over 20 standards covering biological evaluation of medical devices. ISO 10993-1 serves as the parent standard, requiring manufacturers to evaluate and test biocompatibility of any material contacting the human body, categorized by contact type and duration.
Key sub-standards include:
- ISO 10993-5:2009 — In vitro cytotoxicity testing (fully FDA-recognized, Rec #2-245)
- ISO 10993-4:2017+Amd1:2025 — Blood interaction testing (fully FDA-recognized, Rec #2-311)
Important limitation: The FDA only partially recognizes ISO 10993-1:2018, noting conflicts with FDA guidance. Manufacturers must consult FDA-specific biocompatibility guidance (September 2023) to bridge these gaps.
IEC 62304 and IEC 62366-1: Software and Usability
IEC 62304:2006+A1:2015 defines lifecycle requirements for medical device software, covering:
- Software development and maintenance processes
- Risk management for software components
- Software as a Medical Device (SaMD)
- Software used in production or quality systems
The FDA fully recognizes this consolidated version (Rec #13-79).
Usability follows a different framework. IEC 62366-1:2015+A1:2020 specifies usability engineering processes to ensure devices are designed with human factors safety in mind. It requires:
- Structured user research and task analysis
- Use-related risk analysis
- Validation testing with representative users
- Documentation of use errors and mitigation strategies
Together, these two standards cover the full scope of software and human-interface risk — areas regulators increasingly scrutinize in submissions.
ISO 11135 and ISO 11137: Sterilization Standards
ISO 11135:2014+A1:2018 covers sterilization using ethylene oxide (EO), a common method for heat-sensitive devices. It specifies requirements for developing, validating, and routinely controlling the sterilization process. The FDA fully recognizes this standard (Rec #14-529).
ISO 11137-1:2025 covers radiation sterilization using Cobalt-60, Cesium-137, electron beams, or X-rays. The FDA recognizes the 2025 edition (Rec #14-611), with a transition period allowing use of the 2006 edition until July 4, 2027.
Manufacturers of sterile medical devices must comply with both — each requires rigorous process validation and ongoing routine monitoring.
How ISO Standards and FDA Regulations Work Together
The FDA's Quality System Regulation (QSR) under 21 CFR Part 820 was first implemented in 1978 under the Federal Food, Drug and Cosmetic Act. After the Safe Medical Devices Act of 1990 (which found that faulty product design contributed significantly to device recalls), the FDA revised Part 820 in 1996 to introduce preproduction design controls and harmonize with ISO 9001 and early drafts of ISO 13485.
The 2024 QMSR Final Rule
On February 2, 2024, the FDA published a final rule amending 21 CFR Part 820, creating the Quality Management System Regulation (QMSR). This regulation becomes effective February 2, 2026, and legally incorporates ISO 13485:2016 by reference.
The QMSR retains FDA-specific requirements to ensure statutory compliance:
- §820.10 links ISO 13485 clauses to FDA rules for Unique Device Identification (UDI), traceability, complaint handling, medical device reporting, and recalls
- §820.45 mandates specific device labeling and packaging controls, including expiration dates and storage instructions

FDA Recognition vs. Legal Equivalence
When the FDA "recognizes" a consensus standard like ISO 14971, manufacturers can submit a Declaration of Conformity as evidence of meeting related FDA requirements, reducing the need for underlying raw data. However:
- Recognition does not replace FDA registration, 510(k) clearance, or PMA approval
- Certificates do not substitute for FDA inspections
- Deviating from recognized normative sections invalidates the Declaration of Conformity
FDA alignment is only part of the picture for companies selling beyond U.S. borders.
Global Market Considerations
Companies distributing or exporting medical devices must also consider destination market requirements:
- EU MDR requires conformance to harmonized EN ISO standards published in the Official Journal of the European Union (OJEU) to claim presumption of conformity
- Health Canada strictly requires Class II, III, and IV device manufacturers to provide ISO 13485 quality system certificates from recognized third-party auditors
Working with a 3PL partner that holds its own ISO and GMP compliance — and understands FDA packaging and labeling requirements — helps protect product conformity at every stage, from storage to final delivery, and reduces the risk of customs delays or market withdrawal.
ISO Standards for Medical Device Packaging and Distribution
Maintaining sterile barrier integrity and proper labeling from manufacturing through final delivery is a critical regulatory requirement that extends beyond the manufacturer's facility.
ISO 11607-1 and ISO 11607-2: Sterile Barrier Systems
- ISO 11607-1:2019+A1:2023 specifies requirements for materials, sterile barrier systems, and packaging systems (FDA Rec #14-594)
- ISO 11607-2:2019+A1:2023 covers validation of forming, sealing, and assembly processes (FDA Rec #14-595)
Both standards are fully FDA-recognized and mandatory for terminally sterilized medical devices. Proper packaging compliance ensures sterility is maintained throughout the entire distribution chain—from the manufacturing floor through warehousing, transportation, and final delivery.
ISO 15223-1: Medical Device Symbols
ISO 15223-1:2021 governs the symbols used on medical device labels and packaging—covering markings such as expiration dates, sterile indicators, and handling instructions. Standardized symbols ensure these markings are understood across international markets, reducing the risk of misuse and customs rejection.
Downstream Compliance Responsibilities
These labeling and packaging standards don't stop applying once a device leaves the manufacturer's dock. Every downstream handler—from warehouse operators to final delivery partners—must uphold the same conditions these standards require.
Under the QMSR, Clause 7.5.11 of ISO 13485 requires manufacturers to preserve device conformity throughout the supply chain. In practice, that means logistics partners need documented procedures for:
- Temperature control and humidity monitoring
- Protection from contamination during storage and transit
- Proper handling to prevent physical damage
- Traceability through each stage of distribution

Working with a logistics provider that holds FDA, ISO, and GMP compliance helps manufacturers keep their regulatory requirements intact beyond their own facility. Bluebonnet Medical Supplies, for instance, offers FDA-cleared medical packaging and GMP-compliant warehousing for medical device distribution—maintaining compliance from storage through final shipment.
Frequently Asked Questions
What are ISO standards for medical devices?
ISO standards are internationally recognized, consensus-based documents developed by the International Organization for Standardization that define quality, safety, and performance requirements for medical devices. While technically voluntary, they are recognized or harmonized by regulators like the FDA and EU, making them essential for market access worldwide.
What is ISO 13485 standard for medical devices?
ISO 13485 is the global standard for Quality Management Systems specific to medical device manufacturers. It specifies the processes, documentation, and controls needed to produce safe, effective devices in compliance with regulatory requirements. It requires third-party certification and ongoing maintenance through surveillance audits.
How does ISO 13485 differ from ISO 9001, ISO 14971, and ISO 15189?
ISO 13485 is the device-specific QMS standard — it incorporates risk management and regulatory compliance as core requirements, not optional add-ons. ISO 9001 is a general QMS for any industry; ISO 14971 covers medical device risk management specifically; ISO 15189 addresses quality in medical laboratories. Each serves a distinct scope.
What are ISO 45001, ISO 14001, and ISO 9001?
ISO 9001 covers general quality management for any industry. ISO 14001 is an environmental management standard. ISO 45001 addresses occupational health and safety. None are medical-device-specific, but device companies may pursue them as part of broader operational compliance.
What is 21 CFR 820 for medical devices?
21 CFR Part 820 is the FDA's Quality System Regulation, recently updated and renamed the Quality Management System Regulation (QMSR). Effective February 2, 2026, it legally incorporates ISO 13485:2016 and sets mandatory requirements covering the full lifecycle of medical devices sold in the US — from design and manufacture through distribution.
What is the difference between ISO 14937 and ISO 11135?
ISO 11135 specifically governs sterilization of medical devices using ethylene oxide (EO). ISO 14937 is a broader standard covering the characterization and development of sterilization processes using other sterilizing agents such as moist heat, dry heat, or chemical methods not covered by specific standards. The choice depends on the sterilization method used.


