
Introduction
Many medical device manufacturers struggle with packaging failures that compromise sterility, trigger regulatory non-conformances during FDA audits, or result in products being held at customs. ISO 11607 addresses these challenges as the internationally recognized standard governing packaging for terminally sterilized medical devices, comprising two parts: Part 1 addresses materials and sterile barrier system requirements, while Part 2 covers validation of forming, sealing, and assembly processes.
For medical device manufacturers, distributors, quality teams, and online medical supply businesses, packaging compliance is a patient safety issue—not just a regulatory one. When sterile barriers fail, the consequences extend well beyond a Form 483 observation. According to FDA enforcement data, packaging-related failures remain a persistent cause of 483 observations and product recalls, with inadequate seal validation and distribution testing among the most frequently cited deficiencies.
This guide covers what each part of ISO 11607 requires, how packaging validation works in practice, and what drives compliance risk across the full supply chain—from manufacturing through final delivery.
TL;DR
- ISO 11607 is the global standard for sterile medical device packaging, recognized by the FDA and aligned with EU MDR
- Part 1 covers materials, sterile barrier systems, and performance requirements
- Part 2 addresses packaging process validation (installation, operational, and performance qualification)
- Compliance requires maintaining packaging integrity through storage, handling, and distribution—not just at manufacture
- The FDA accepts declarations to the 2019 versions (without 2023 amendment) only until December 20, 2026
- Non-compliance risks product recalls, customs holds, and patient safety incidents
What Is ISO 11607?
ISO 11607 is a two-part international standard that specifies requirements for packaging systems used with terminally sterilized medical devices—devices that are sterilized inside their final packaging. The standard ensures a device reaches the point of use in a sterile, undamaged condition by establishing strict requirements for packaging materials, design, and manufacturing processes.
Current versions:
- ISO 11607-1:2019 (confirmed 2024): Requirements for materials, sterile barrier systems, and packaging systems
- ISO 11607-2:2019 (confirmed 2024): Validation requirements for forming, sealing, and assembly processes
- Amendment 1:2023: Both parts received a critical amendment integrating risk management requirements directly into the packaging lifecycle
ISO 11607 is not a general packaging standard. Where most packaging specs focus on aesthetics or transit durability, this standard governs microbial barrier performance, seal integrity, and process controls—because contamination in sterile medical packaging directly affects patient safety. Its requirements follow the packaging system all the way through end use, not just off the production line.
Why ISO 11607 Matters for Medical Device Packaging
Dual Regulatory Recognition
ISO 11607 holds unique regulatory status:
- FDA Recognition: The FDA recognizes ISO 11607-1:2019 and ISO 11607-2:2019 including AMD1:2023 as consensus standards. Declarations of conformity to the 2019 versions without the 2023 amendment will only be accepted until December 20, 2026
- EU Harmonization: The European Commission harmonized EN ISO 11607-1:2020/A1:2023 and EN ISO 11607-2:2020/A1:2023 under MDR 2017/745 in March 2024
This dual recognition makes compliance effectively mandatory for market access in both the US and EU.
Real-World Consequences of Non-Compliance
When packaging fails to meet ISO 11607 requirements, consequences extend beyond regulatory citations:
Regulatory enforcement: FDA Form 483 observations frequently cite ISO 11607-2 failures. A published 2016 FDA 483 response for Zimmer Biomet highlighted failures in Operational and Performance Qualifications for sealers, specifically noting that seal integrity verification was inadequate. PQ studies also failed to challenge the process with expected manufacturing variables, such as power failures.
Product recalls: FDA recall data shows a persistent trend of design failures where devices puncture pouches during transit because worst-case distribution simulation testing was either not performed or didn't utilize the heaviest or sharpest device configurations.
Customs holds: Products shipped internationally can be stopped at customs when packaging documentation doesn't demonstrate conformity to recognized standards, delaying delivery and creating costly compliance corrections.
Patient safety risks: Compromised sterile barriers create direct contamination risks that can result in surgical site infections or device-related adverse events.

Ongoing Compliance Responsibility
ISO 11607 is not a one-time checkbox. Packaging must be re-validated whenever key conditions change. Common re-validation triggers include:
- Material or supplier changes (films, lids, adhesives)
- New or modified sterilization methods
- Device configuration changes (size, weight, sharp features)
- Updated distribution routes or environmental conditions
- Significant process equipment changes or moves
Each trigger requires documented change control records and updated validation studies to maintain regulatory standing.
ISO 11607-1: Requirements for Packaging Materials and Sterile Barrier Systems
The Two-Layer Packaging Configuration
ISO 11607-1 requires a two-layer approach to sterile medical packaging.
The sterile barrier system (SBS) is the minimum package that provides a microbial barrier, prevents contamination, and allows aseptic presentation at the point of use. Protective packaging surrounds the SBS to prevent physical or environmental damage during handling, storage, and transit. Both layers must be validated together as a complete packaging system.
Common Sterile Barrier System Formats
ISO 11607-1 references several SBS formats:
- Peel pouches with medical-grade paper and transparent film
- Sterilization bags and wraps
- Rigid trays with die-cut or thermoformed lids
- Form/fill/seal systems created inline during production
- Four-side-seal configurations for flat devices
The standard distinguishes "preformed sterile barrier systems" (supplied partially assembled for filling at healthcare facilities) from fully assembled formats used in commercial device packaging.
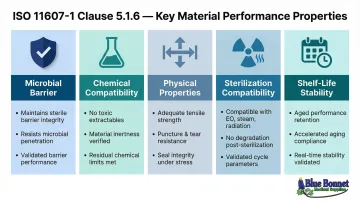
Five Key Performance Properties
ISO 11607-1 Clause 5.1.6 mandates evaluation of specific material properties. Testing each of these properties requires accepted standardized methods — but the requirements themselves define what each material must actually demonstrate.
| Property | Key Requirements |
|---|---|
| Microbial barrier | Porous materials tested for bacterial spore blocking; impermeable materials evaluated via air permeance; ingress prevention required throughout shelf life |
| Chemical | Must meet pH and sulfate specifications; must not release hazardous substances; device material compatibility must be demonstrated |
| Physical | Free of defects (holes, cracks, delamination); meets tensile strength, tear resistance, and burst strength specs; seal areas require peel strength or integrity testing |
| Sterilization compatibility | Must allow full sterilization of the enclosed device; must not produce excessive residues (such as ethylene oxide retention); validation is method-specific |
| Shelf-life stability | All properties must remain within limits over shelf life; temperature, humidity, and light exposure must be accounted for; accelerated aging studies (typically per ASTM F1980) support expiration date claims |
Standardized Test Methods
Annex B of ISO 11607-1 lists accepted test methods from ISO, ASTM, CEN (EN), and TAPPI — over 100 in total, covering accelerated aging, seal strength, package integrity, and microbial barrier performance.
Manufacturers may use other methods with proper justification, providing some flexibility in test selection based on specific packaging configurations.
Special Requirements for Reusable Containers
Clauses 5.1.10 and 5.1.11 establish additional requirements for reusable sterile barrier systems:
- Must be evaluated for degradation over their service life
- Must be labeled with maximum reuse counts or visual inspection acceptance criteria
- Must feature tamper-evident systems
- Sterilizing agent ports must provide a microbial barrier when closed
ISO 11607-2: Validation Requirements for Packaging Processes
What ISO 11607-2 Covers
ISO 11607-2 addresses how packaging is manufactured, not just what it's made of. Forming, sealing, and assembly processes used to create the sterile barrier system must be formally validated — confirming they reliably produce packaging that meets all ISO 11607-1 requirements.
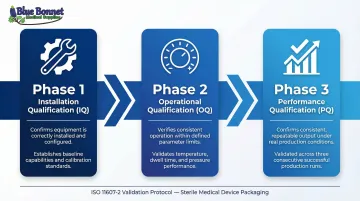
Three-Phase Validation Protocol
Clause 5.1.2 requires that process validation include:
Installation Qualification (IQ):
- Confirms equipment is installed correctly
- Verifies equipment meets manufacturer specifications
- Documents baseline equipment capabilities before production use
Operational Qualification (OQ):
- Verifies equipment operates consistently within defined process parameters
- Challenges process variables to determine upper and lower parameter limits
- Tests critical parameters such as heat sealer temperature, dwell time, and pressure
- Establishes the validated operating range for routine production
Performance Qualification (PQ):
- Confirms the validated process consistently produces packaging meeting ISO 11607-1 requirements
- Conducted under real production conditions: machine setup, operator changes, and shift variations
- Requires at least three production runs to demonstrate consistency
- Includes worst-case challenges such as startup conditions or maximum production speeds
Monitoring Production After Validation
Under Clause 5.6, manufacturers must establish procedures to keep the process under control during routine production. Specified process variables must be monitored and recorded, with documented acceptance criteria and corrective action steps when parameters drift outside validated ranges.
Triggers for Revalidation
Clause 5.7.2 identifies specific triggers requiring revalidation:
- Raw material changes that impact process variables
- Equipment modifications or transfers to new locations
- Changes to process parameters outside the validated range
- Negative trends in quality control indicators
- Changes in production volumes that affect process consistency
How Part 1 and Part 2 Work Together
ISO 11607-1 defines what the final packaging must achieve; ISO 11607-2 ensures the manufacturing process reliably gets you there. Full compliance requires both — meeting material requirements alone won't satisfy regulators if the production process hasn't been formally validated.
Key Factors That Affect ISO 11607 Compliance
Sterilization Method Dependency
Different sterilization methods place different stresses on packaging materials:
Ethylene oxide (EO): Requires porous materials for gas penetration; materials must not retain excessive EO residues; relatively gentle on most polymers.
Gamma radiation: Can cause polymer degradation, discoloration, or embrittlement; requires materials specifically validated for radiation doses used.
Steam autoclaving: Subjects packaging to high temperature and moisture; requires materials that maintain seal integrity under wet heat conditions.
Critical compliance point: A packaging system validated for EO sterilization is not automatically valid for gamma radiation. Each sterilization method requires separate validation studies demonstrating that the SBS maintains all required properties after exposure.
Distribution and Handling Conditions
Packaging that passes factory validation can still fail in the field if distribution and handling conditions exceed validated parameters:
Physical stress: Compression from stacking, vibration during transit, and impact from handling can compromise seal integrity or puncture barrier materials.
Environmental exposure: Humidity variation during warehousing, temperature extremes during shipping, and UV light exposure can degrade packaging materials over time.
Compliance chain: Validated packaging integrity only holds if every downstream handler meets the same standards. A 3PL partner like Bluebonnet Medical Supplies that operates under FDA-cleared, ISO- and GMP-compliant conditions helps ensure that proper storage, careful handling, and fulfillment checks preserve sterile barrier integrity all the way to the end user.
Even with a compliant logistics partner in place, time itself becomes a compliance variable — which is where shelf-life testing enters the picture.
Shelf-Life Testing Requirements
ASTM F1980-21 is the FDA-recognized standard for accelerated aging studies. These studies model the effects of time on sterile barrier integrity to support expiration date claims until real-time aging data becomes available.
Key considerations:
- Accelerated aging conditions must reflect expected storage environments
- Temperature and humidity variation must be accounted for in study design
- Packaging must be tested for all required properties after accelerated aging
- Real-time aging studies must eventually confirm accelerated aging predictions
Shelf-life data tells you whether packaging holds up over time — but seal integrity testing confirms whether each individual package is actually sealed correctly in the first place.
Seal Integrity Testing
Seal integrity is one of the most closely scrutinized compliance points during regulatory audits.
ISO 11607 does not specify a single universal seal strength value—instead, manufacturers must establish and validate their own minimum and maximum acceptable seal strength limits.
Common seal integrity test methods:
| Test Method | Application | Detection Capability |
|---|---|---|
| ASTM F88/F88M-23 | Peel strength testing for flexible pouches | Quantitative measure of force required to separate seal |
| ASTM F1929-23 | Dye penetration for porous materials | Detects and locates channels ≥ 50 µm in edge seals |
| ASTM F2096-11 | Bubble emission testing | Detects gross leaks down to 250 μm in trays and pouches |
| ASTM F2338-24 | Vacuum decay (non-destructive) | Highly sensitive; suitable for 100% online production testing |

The acceptable seal strength range must be established during OQ/PQ validation and documented as part of the validation package.
Common Misconceptions About ISO 11607
"Validation Is a One-Time Event"
One of the most frequent compliance failures is the assumption that passing ISO 11607 testing once is sufficient. In reality, validation must be repeated or bridged whenever there is a change to:
- Materials or suppliers
- Equipment settings or transfers
- Device configuration or packaging design
- Sterilization method or parameters
- Distribution routes or expected storage conditions
Maintaining compliance means establishing documented change control procedures and scheduling periodic reviews to confirm the validated state holds over time.
"The Outer Carton Is the Sterile Barrier System"
Manufacturers sometimes mistake the outer carton or corrugated shipper for the validated SBS. Annex E of ISO 11607-1 specifically addresses this confusion: only the inner layer meeting microbial barrier requirements qualifies as the SBS.
SBS vs. protective packaging:
- The SBS provides the microbial barrier and enables aseptic presentation
- Protective packaging shields the SBS from physical or environmental damage
- Both layers serve distinct functions and must be clearly labeled to differentiate them
Introducing unvalidated protective packaging into a sterile field — whether through mislabeling or user error — creates a direct contamination risk.
"Compliance Ends at the Factory Door"
ISO 11607 applies to the entire packaging system through the point of use — not just at the point of manufacture. A seal that passes post-production inspection can still represent a compliance failure if distribution and storage conditions fall outside the parameters assumed during validation.
What this means in practice:
- Distribution simulation testing must reflect actual shipping conditions
- Warehouse storage must maintain temperature and humidity within validated ranges
- Handling procedures must prevent physical damage to validated packaging
- Documentation must demonstrate control of conditions throughout the supply chain
Frequently Asked Questions
What is ISO 11607?
ISO 11607 is an international standard specifying requirements for packaging systems used with terminally sterilized medical devices. Part 1 covers materials and sterile barrier system requirements; Part 2 covers process validation requirements. It is recognized by the FDA and harmonized under the EU MDR.
What is ISO 11607-1?
ISO 11607-1 covers the requirements for packaging materials, sterile barrier systems, and the complete packaging system—including properties such as microbial barrier, physical integrity, chemical compatibility, sterilization compatibility, and shelf-life stability.
What is ISO 11607-2?
ISO 11607-2 covers the validation requirements for the processes used to form, seal, and assemble sterile packaging. It requires IQ, OQ, and PQ validation of sealing and forming equipment, plus ongoing process monitoring in routine production.
What is the latest version of ISO 11607-1?
The current version is ISO 11607-1:2019 (published February 2019, confirmed 2024). It received an amendment in September 2023 (ISO 11607-1:2019/Amd 1:2023) that integrated risk management requirements directly into the standard. The FDA will accept declarations to the 2019 version without the amendment only until December 20, 2026.
What is the ISO 11607 requirement for seal strength?
ISO 11607 does not specify a single universal seal strength value. Instead, manufacturers must establish and validate their own minimum and maximum acceptable seal strength limits through testing (e.g., peel strength testing per ASTM F88). These limits must be documented as part of the process validation package.


