
Introduction
Medical device packaging failures don't just mean damaged products — they mean compromised sterility, patient safety risks, regulatory violations, and costly product recalls. According to FDA databases, seal integrity failures alone account for nearly 800 medical device recalls between 2019 and 2025, with 96.7% of those seal-related recalls involving medical devices. The Exactech recall — caused by packaging missing an oxygen barrier layer — led to accelerated implant degradation and contributed to the company filing for bankruptcy.
If you manufacture, sell, or ship medical devices — domestically or internationally — packaging compliance isn't optional. The requirements are specific, the consequences of non-compliance are severe, and the regulations governing them are changing.
Key Takeaways:
- Packaging is a regulated device component under FDA 21 CFR Part 820 and ISO 11607 — not just protective material
- FDA's new QMSR (effective Feb 2026) requires documented pre-release packaging inspections
- Sterile devices require validated Sterile Barrier Systems with real-time aging studies
- UDI labeling is mandatory on all device packages under 21 CFR Part 801.20
- Package validation requires IQ/OQ/PQ qualification and integrity testing per ASTM standards
What Is Medical Device Packaging and Why It Matters
Medical device packaging encompasses the complete system used to contain, protect, and preserve a device from manufacture through to the end user. Three layers make up this system:
- Primary packaging: Direct contact with the device (sterile barrier systems, pouches, trays)
- Secondary packaging: Outer cartons or boxes that group primary packages
- Tertiary packaging: Shipping containers and pallets for bulk transport
Why Packaging Is a Regulated Device Component
Under 21 CFR Part 820.1, FDA states that current good manufacturing practice (CGMP) requirements govern the "design, manufacture, packaging, labeling, storage, installation, and servicing of all finished devices." This means packaging failures carry the same regulatory weight as mechanical or software failures in the device itself.
Real-world consequences include:
- Advanced Sterilization Products paid a $1.25 million FDA civil penalty for unsupported shelf-life claims on packaging
- Integra LifeSciences issued a Class II recall of MediHoney products over sterile barrier breaches, resulting in 11 serious injuries
- Exactech's defective packaging recall contributed directly to the company's bankruptcy filing

Sterile vs. Non-Sterile Device Packaging
Sterile devices carry far more regulatory burden than non-sterile devices:
Sterile device requirements:
- Validated Sterile Barrier System (SBS) per ISO 11607
- Package integrity testing (dye penetration, bubble testing)
- Seal strength validation (peel testing)
- Accelerated and real-time aging studies
- Sterilization method compatibility verification
Non-sterile devices face a lighter compliance load, though labeling and distribution requirements still apply:
Non-sterile device requirements:
- Protective packaging to maintain product integrity
- Labeling compliance under 21 CFR Part 801
- Distribution simulation testing
- Basic quality control inspections
Key Regulatory Standards for Medical Device Packaging
FDA's Quality Management System Regulation (QMSR)
On February 2, 2024, FDA published the final QMSR, effective February 2, 2026. This regulation harmonizes FDA requirements with ISO 13485:2016 and transitions packaging requirements from 21 CFR 820.130 to § 820.45.
FDA noted that ISO 13485 Clause 7.5.1(e) lacks sufficient specificity for packaging inspections, so § 820.45 mandates that manufacturers:
- Inspect labeling and packaging for accuracy before release
- Verify UDI, expiration dates, and storage instructions
- Document all inspections to prevent mix-ups
ISO 11607: The Global Packaging Standard
ISO 11607 is the internationally recognized consensus standard for terminally sterilized medical device packaging. FDA formally recognizes the current versions and will accept declarations of conformity to the 2019 versions only until December 20, 2026.
ISO 11607-1:2019 (Including AMD1:2023) covers the materials side: requirements for sterile barrier systems, packaging materials, and the test methods needed to confirm sterility is maintained until point of use.
ISO 11607-2:2019 (Including AMD1:2023) addresses process validation — specifically forming, sealing, and assembly — and mandates the IQ/OQ/PQ qualification framework.
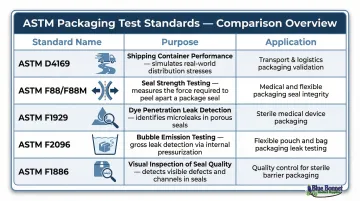
ASTM Testing Standards
FDA recognizes specific ASTM standards for package testing and validation:
| ASTM Standard | Purpose | Application |
|---|---|---|
| ASTM D4169 | Performance testing of shipping containers | Simulates distribution hazards (drops, vibration, compression) |
| ASTM F88/F88M | Seal strength testing | Measures force required to separate seals |
| ASTM F1929 | Dye penetration testing | Detects channel leaks in edge seals |
| ASTM F2096 | Bubble emission testing | Detects gross leaks through internal pressurization |
| ASTM F1886 | Visual inspection of seals | Identifies visual seal defects |
EU MDR and International Requirements
EU Medical Device Regulation (MDR) 2017/745 imposes strict packaging requirements under Annex I General Safety and Performance Requirements:
- GSPR 11.4: Sterile device packaging must ensure sterility under specified transport and storage conditions, with integrity "clearly evident to the final user"
- GSPR 11.7: Non-sterile device packaging must maintain product integrity and minimize microbial contamination risk
For international markets, additional requirements may include CE marking, translated labeling, and market-specific symbol standards beyond FDA requirements.
Design History File and Technical Documentation
FDA requires packaging validation to be documented in:
- Design History File (DHF) per 21 CFR 820.30(j)
- Process validation records per 21 CFR 820.75
EU MDR carries its own parallel documentation burden. Annex II requires the Technical Documentation to include complete manufacturing process information, validation reports for packaging and sterilization, bioburden testing, and sterility maintenance documentation.
FDA Labeling Requirements for Medical Device Packaging
Mandatory Label Elements Under 21 CFR Part 801
FDA labeling requirements fall under 21 CFR Part 801. Non-compliant labeling renders a device misbranded, which triggers FDA enforcement action.
Required label elements:
- Name and place of business of manufacturer, packer, or distributor (including street address, city, state, ZIP code)
- Accurate statement of quantity of contents
- Directions for use (if not self-evident)
- Adequate warnings or precautions
- Device's intended use (if not self-evident)
Note: Expiration and manufacturing dates must follow the exact format YYYY-MM-DD (e.g., 2024-03-15). Incorrect date formatting alone is enough to render a device misbranded.
Unique Device Identifier (UDI) Requirements
Under 21 CFR Part 830, most medical devices must carry a UDI on their labels and be registered in FDA's GUDID database. Every device label and package must bear a UDI in both plain-text and Automatic Identification and Data Capture (AIDC) technology.
UDI components:
- Device Identifier (DI): Fixed portion identifying the specific version/model and labeler
- Production Identifier (PI): Variable portion identifying lot/batch, serial number, expiration date, and/or manufacturing date
Compliance timeline by device class:
- Class III: September 24, 2014
- Implantable/Life-Supporting: September 24, 2015
- Class II: September 24, 2016
- Class I: September 24, 2018
Sterile Device Labeling Requirements
Packaging for sterile devices must clearly indicate:
- "STERILE" marking (not optional)
- Sterilization method used
- Expiration date or shelf life of sterility
- Confirmation that packaging integrity has been maintained (required for international shipments under EU MDR GSPR 23.3)
- Instructions on what to do if sterile packaging is damaged
ISO 15223-1 Symbols
FDA recognizes ISO 15223-1:2021 (including AMD1: 2025) for standardized medical device symbols. Rather than printing explanatory text next to each symbol on the label, manufacturers can use the symbols alone — as long as the device labeling includes a complete symbols glossary (paper or electronic).
Two practical points worth knowing:
- Symbols reduce label clutter and support international readability
- The glossary must be included in labeling, but doesn't need to appear on the physical package itself
Types of Medical Device Packaging
Primary, Secondary, and Tertiary Packaging Levels
Primary Packaging (Sterile Barrier System):
- Minimum package that prevents microorganism ingress
- Allows aseptic presentation at point of use
- Examples: Tyvek pouches, thermoformed trays, blister packs
Secondary Packaging (Protective Packaging):
- Outer cartons or boxes grouping primary packages
- Provides cushioning and distribution protection
- Must be validated but serves different function than SBS
Tertiary/Shipping Packaging: Corrugated boxes and pallets used for bulk transport, protecting multiple secondary packages throughout distribution.
Common Packaging Formats
- Tyvek/foil/film flexible pouches — breathable for EO sterilization or impermeable for radiation sterilization
- Thermoformed rigid trays with lidding materials for structural protection and aseptic presentation
- Clamshells made from two-piece rigid plastic with hinged or separate components
- Bottles and vials for liquid-containing devices or components requiring sealed containers
Sterilization Method Compatibility
Choosing the right format means nothing if the material can't survive sterilization. Every packaging decision must align with the sterilization method used:
| Sterilization Method | Material Requirements | Compatible Materials |
|---|---|---|
| Ethylene Oxide (EO) | Breathable/permeable to allow gas ingress and egress | Tyvek/Mylar combinations, Tyvek/PETG trays, medical-grade paper |
| Radiation (Gamma/E-beam) | Can be impermeable; must resist radiation-induced degradation | Foils, Mylar, impermeable polymer films |
| Moist Heat (Steam) | Must allow steam penetration; withstand high temperature and pressure | Paper/plastic pouches, high-temp Tyvek/film combinations |
Critical error: Selecting an impermeable foil pouch for an EO-sterilized device will result in complete sterilization failure.
Package Testing and Validation Requirements
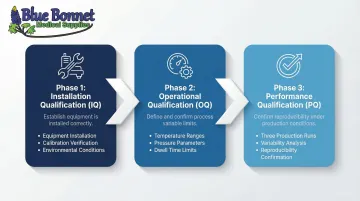
The IQ/OQ/PQ Validation Framework
Package validation is mandatory before commercial distribution of sterile medical devices. ISO 11607-2 requires a three-phase equipment qualification process:
1. Installation Qualification (IQ):
- Documents that equipment is installed correctly
- Records equipment design features, safety features, environmental conditions, and calibration schedules
2. Operational Qualification (OQ):
- Challenges process variables to determine upper and lower parameter limits
- Establishes temperature, pressure, and dwell time ranges that produce acceptable sterile barrier systems
3. Performance Qualification (PQ):
- Demonstrates consistent production of acceptable packaging under normal operating conditions
- Requires minimum of three production runs to assess variability and reproducibility
Package Integrity Testing
Integrity testing verifies there are no defects — pinholes, channel leaks, delamination — that could compromise sterility.
Accepted test methods:
- ASTM F1929 (dye penetration) — identifies channel leaks in edge seals by applying a dye penetrant to the seal area
- ASTM F2096 (bubble testing) — pressurizes the package internally while submerged to detect gross leaks
- ASTM F1886 (visual inspection) — flags seal defects that point to heat sealing process variations
Package Strength Testing
Seal strength testing quantifies the force required to open the seal and ensures packages survive distribution without failing.
Primary method:
- ASTM F88/F88M (peel testing) — measures the force required to separate a seal, confirming it holds during transit while still opening cleanly during aseptic presentation
Test types:
- Destructive tests (peel, burst) consume the sample but provide direct strength measurements
- Non-destructive tests (visual inspection, certain bubble tests) keep the package intact for continued use
Distribution Simulation and Aging Studies
Distribution testing (ASTM D4169):
- Simulates full supply chain lifecycle
- Includes drop testing, vibration, compression, and environmental conditioning
- Required before regulatory submission
Distribution testing tells you whether the package survives the journey — aging studies tell you whether it survives time.
Aging studies:
- Accelerated aging (ASTM F1980) uses elevated temperatures to simulate the effects of time on sterile integrity
- Real-time aging runs parallel studies at normal storage conditions to confirm the accelerated data
FDA and ISO 11607 accept accelerated aging for initial market-entry shelf-life claims, but both require parallel real-time aging studies to confirm those results. If real-time data fails to corroborate accelerated findings, shelf-life must be reduced — and products may face recall.
Common Compliance Mistakes and How to Avoid Them
Most Frequent Packaging Compliance Errors
Starting packaging design too late:
- Packaging should be part of initial device development, not an afterthought
- Late-stage redesigns are costly and delay market entry
Failing to validate packaging changes:
- Any change to materials, sealing parameters, or processes requires revalidation
- Even "minor" changes can compromise sterile barrier integrity
Neglecting label updates:
- Labels must be updated whenever device specifications, intended use, or regulatory requirements change
- Outdated labels render devices misbranded
Inadequate documentation:
- FDA inspections require complete Design History Files and process validation records
- Missing documentation can result in warning letters even if packaging performs correctly
Real-World Consequences
FDA enforcement data points to two consistent patterns:
- A retrospective analysis of FDA warning letters from 2010 to 2020 found that CGMP non-compliance (particularly validation failures) and misbranding — including labeling and packaging errors — were the top two reasons for issuance
- FY2017 FDA data showed Production and Process Controls, which include packaging requirements, accounted for 37% of all Warning Letter citations
Financial impact examples:
- Regulatory penalties up to $1.25 million for packaging-related violations
- Product recalls requiring logistics coordination and customer notification
- Customs rejection for international shipments
- Product liability lawsuits from packaging failures causing patient harm
Working with a Qualified 3PL Partner
The financial and operational risks above are preventable — but only if compliance is built into your packaging process from the start, not patched in after a warning letter arrives.
Companies without in-house expertise to manage packaging compliance throughout the process can work with a qualified 3PL partner holding FDA, ISO, and GMP credentials. Bluebonnet Medical Supplies integrates compliance checks directly into daily warehouse operations — covering sterile packaging, labeling accuracy, and storage conditions. Products are packed, stored, and shipped in full accordance with regulatory requirements, reducing the risk of customs holds and compliance failures without requiring businesses to build that infrastructure themselves.

Frequently Asked Questions
What is medical device packaging?
Medical device packaging refers to the complete system of materials, containers, and sterile barriers used to protect a medical device from manufacture through to the end user. It is a regulated component of the device subject to FDA 21 CFR Part 820 and international standards like ISO 11607.
What are the different types of medical device packaging?
There are three levels: primary packaging (sterile barrier systems like Tyvek pouches and thermoformed trays), secondary packaging (outer cartons for distribution protection), and tertiary packaging (shipping containers and pallets for bulk transport). Common formats include flexible pouches, rigid trays with lidding, and clamshells.
What are the packaging standards for medical devices?
Key standards include ISO 11607 for terminally sterilized devices, FDA's 21 CFR Part 820 (transitioning to QMSR in 2026), and ASTM testing standards such as D4169, F88, and F1929. ISO 11607 is recognized by FDA as a consensus standard.
What are the FDA requirements for medical device labeling?
21 CFR Part 801 requires manufacturer identification, quantity of contents, directions for use, and adequate warnings. 21 CFR Part 830 mandates UDI compliance for most device classes, displayed in both plain-text and AIDC formats, with dates in YYYY-MM-DD format.
What happens if medical device packaging doesn't meet requirements?
Non-compliant packaging can trigger FDA warning letters, product recalls, import alerts, and customs rejections. It can block a product from market entirely and expose manufacturers to civil penalties exceeding $1 million.
Do medical device packaging requirements differ for international shipments?
Yes, international markets have their own packaging and labeling standards. The EU under MDR 2017/745 requires additional documentation, CE marking, and specific sterile packaging declarations. Other global markets may require translated labeling, different symbol sets, or conformity assessments beyond what FDA requires domestically.


